




What is Next Generation Sequencing (NGS)?
Next-generation sequencing (NGS), is a sequencing technique that allows for extremely fast, scalable, and high-throughput sequencing. It is used to identify the sequence of nucleotides in targeted areas of DNA or RNA or even entire genomes.
The development of NGS has transformed biological research, providing unprecedented opportunities to study biological systems and perform a diverse range of applications. Traditional DNA sequencing technologies are insufficient for answering the complex questions in genomics today, and NGS has taken their place as an essential tool for addressing these challenges.
But why NGS? you may ask, why not just name it "New Sequencing Technology"?
Next-generation sequencing (NGS) is called so because it represents the next step in the evolution of DNA sequencing technologies beyond the traditional Sanger sequencing method, which was the first generation of DNA sequencing.
NGS platforms were developed in the 2000s, and they have revolutionized the field of genomics by offering ultra-high throughput, scalability, and speed. They can sequence multiple DNA fragments simultaneously, producing large amounts of data in a short amount of time. This has allowed researchers to tackle complex genomic questions that were not possible before, leading to discoveries and insights.
Therefore, NGS is often referred to as the "next generation" of sequencing technologies because of its significantly improved capabilities compared to earlier methods.

Older sequencing technologies
Sanger sequencing: A method for DNA sequencing that uses chain-terminating dideoxynucleotides to generate labelled fragments of DNA.
Maxam-Gilbert sequencing: A method for DNA sequencing that uses chemical cleavage to generate labelled fragments of DNA.
Next-generation sequencing technologies
Pyrosequencing: A method for DNA sequencing that uses a series of enzymatic reactions to generate light signals that are detected by a camera.
Illumina sequencing: A method for DNA sequencing that uses reversible dye-terminator chemistry and massively parallel sequencing-by-synthesis to generate high-throughput, accurate data.
Ion Torrent sequencing: A method for DNA sequencing that uses semiconductor sequencing technology to detect changes in pH as nucleotides are incorporated into a growing DNA strand.
Pacific Biosciences (PacBio) sequencing: A method for DNA sequencing that uses single-molecule, real-time sequencing to generate long reads of DNA.
Oxford Nanopore sequencing: A method for DNA sequencing that uses nanopore technology to measure changes in electrical current as nucleotides pass through a membrane, allowing for real-time sequencing of DNA and RNA.
Note that the boundaries between "older" and "next-generation" sequencing technologies can be somewhat arbitrary, as there is often overlap between the development and use of different sequencing methods. Nonetheless, the technologies listed above can broadly be classified into these two categories based on their historical development and the general nature of their methods.
Here is a table for comparison:
| Features | Sanger Sequencing | Maxam-Gilbert Sequencing | Pyrosequencing | Illumina Sequencing | Ion Torrent Sequencing | Oxford Nanopore Sequencing |
| Date of discovery | 1977 | 1976 | 1996 | 2006 | 2010 | 2014 |
| Speed (X) | 1X | 1X | 5-10X | 10-100X | 5-10X | 5-10X |
| Throughput | Low | Low | Lower than Illumina | High | Lower than Illumina | Lower than Illumina |
| Data output | Limited | Limited | Lower than Illumina | Massive | Lower than Illumina | Massive |
| Cost | Expensive | Expensive | Higher than Illumina | Less expensive | Less expensive | More expensive |
| Coverage | Low (0.5X-1X) | Low (0.5X-1X) | Lower than Illumina (0.1X-0.5X) | High (30X-100X) | Lower than Illumina (0.1X-0.5X) | Lower than Illumina (10X-30X) |
| Error rate | 0.1-1% | 0.1-1% | 0.2-1.6% | 0.1-1% | 1-2% | 5-15% |
| Sample requirements | High | High | Lower than Illumina | Low | Low | Low |
| Read length | long (800-1000 bp) | Short (100-500 bp) | Short (200-400 bp) | Short (up to 300 bp) | Short (up to 400 bp) | Long (up to 2 Mb) |
| Scalability | Limited | Limited | Highly scalable | Highly scalable | Highly scalable | Highly scalable |
| Applications | Limited | Limited | Limited | Diverse | Diverse | Diverse |
Note that the specific values in the table will depend on various factors such as the specific technology, the sequencing platform, and the sequencing mode used, as well as improvements and updates made to each technology over time. Nonetheless, the table provides a general comparison of the key features and historical development of these sequencing technologies.
Behind The Scene (BTS)
The steps before sequencing i.e. steps from sample preparation to data analysis involve:
Sample collection: The first step in NGS sequencing is to collect a biological sample from the organism of interest. This could be anything from blood or tissue samples to environmental samples like soil or water.
DNA or RNA extraction: Once the sample is collected, DNA or RNA is extracted from the sample using various extraction methods. The extracted DNA or RNA needs to be of high quality and purity to ensure accurate sequencing results.
Library preparation: Next, the DNA or RNA is fragmented into smaller pieces and adapters are ligated onto the ends of the fragments. These adapters allow the fragments to be attached to a sequencing platform and are used to identify the sample during data analysis.
Quality control: Before proceeding to sequencing, the quality and quantity of the library is checked using various quality control methods. This ensures that the library is suitable for sequencing and that the sequencing data will be of high quality.

The prepared library is then loaded onto the sequencing platform and the sequencing run is initiated. The sequencing platform reads the sequence of the DNA or RNA fragments, generating large amounts of raw sequencing data.
Here is a short video of Illumina sequencing by synthesis
Overall, NGS sequencing involves a complex and multi-step process that requires expertise in both laboratory techniques and bioinformatics analysis.
Applications of NGS
NGS sequencing is a versatile tool that can be applied to a wide range of biological questions, making it an essential tool for modern biological research.
Whole genome sequencing: Determines the entire DNA sequence of an organism's genome.
Targeted resequencing: Sequencing of specific genomic regions of interest.
De novo sequencing: Reconstruction of an organism's genome without a reference sequence.
RNA-Seq: Sequencing of RNA molecules to determine gene expression levels and transcriptome composition.
ChIP-Seq: Identification of protein-DNA interactions and genome-wide mapping of histone modification patterns.
Metagenomics: Study of microbial communities in different environments, including soil, water, and the human gut.
Epigenomics: Investigation of epigenetic modifications, such as DNA methylation and histone modification patterns.
Structural variant analysis: Detection of large-scale genomic rearrangements, such as deletions, duplications, and inversions.
Single-cell sequencing: Analysis of the genetic information of individual cells, providing insights into cell heterogeneity and development.
Microbial genomics: Investigation of the genetics and evolution of microbial populations, including bacteria, viruses, and fungi.
Long-read sequencing: Generation of reads longer than 10,000 base pairs, enabling genome assembly and resolving structural variation.
Transcriptome assembly: Reconstruction of the complete set of transcripts from RNA-Seq data.
Metatranscriptomics: Analysis of the transcriptional activity of microbial communities in different environments.
miRNA and small RNA analysis: Identification and quantification of small RNA molecules, such as miRNAs, piRNAs, and siRNAs.
Long-range amplicon sequencing: Sequencing of large amplicons, useful for haplotype phasing and detecting structural variants.
HLA typing: Identification of human leukocyte antigen (HLA) alleles, important for transplantation and disease association studies.
Tumor profiling: Detection of somatic mutations and alterations in gene expression levels in cancer cells.
Immunogenomics: Investigation of the genetics and diversity of the immune system, including T cell receptors and B cell receptors.
Forensic DNA analysis: Identification of individuals based on DNA profiles, used in criminal investigations and paternity testing.
Environmental sequencing: Investigation of the microbial diversity in various environments, including oceans, soil, and air.
Each of these applications has unique sample preparation, library construction, sequencing, and bioinformatics analysis requirements.
Types Of NGS Data
Straight out of the sequencers we get the following data formats:
FASTQ: A text-based file format that contains nucleotide sequences and their corresponding quality scores.
BCL: Base Call file format generated by Illumina sequencers that contain raw signal intensities from the sequencing process.
SFF: Standard Flowgram Format used by Roche/454 sequencers that stores nucleotide sequence information as flowgrams.
CEL: Cell intensity file format used by Affymetrix microarray-based sequencers.
ABI: Raw data file format used by ABI sequencers that contain raw signal intensities.
FSA: File format used by Applied Biosystems sequencers that contain raw sequencing data.
ZTR: File format used by Solexa/Illumina sequencers that store raw signal intensities and base calls.
ARK: Raw data file format used by Helicos sequencers that contains nucleotide sequence information.
HDF5: Hierarchical Data Format version 5 used by PacBio sequencers to store raw data.
Most of the time, the data received from NGS sequencers is in the FASTQ file format. In this series, we will be discussing how to analyze and interpret data from the FASTQ format.
How To Analyse NGS Data
Well that is exactly what we will cover in the coming articles in this series, but for reference here are some simplified steps:
Quality control: This involves assessing the quality of the raw data generated by the sequencer, including checking for sequence accuracy, read length, and base quality.
Pre-processing: This includes steps such as filtering out low-quality reads, trimming adapter sequences, and removing contaminants.
Alignment: In this step, the pre-processed reads are aligned to a reference genome or assembled de novo.
Variant calling: This involves identifying differences between the aligned reads and the reference genome, such as SNPs, indels, and structural variants.
Annotation: This involves adding functional information to the identified variants, such as their location within genes, their impact on gene function, and their association with diseases.
Interpretation: Finally, the annotated variants are analyzed to gain insights into the biological system being studied, such as identifying potential drug targets or understanding disease mechanisms.
There are many software tools available to perform these steps, and the specific analysis pipeline used depends on the research question and the type of data being analyzed.
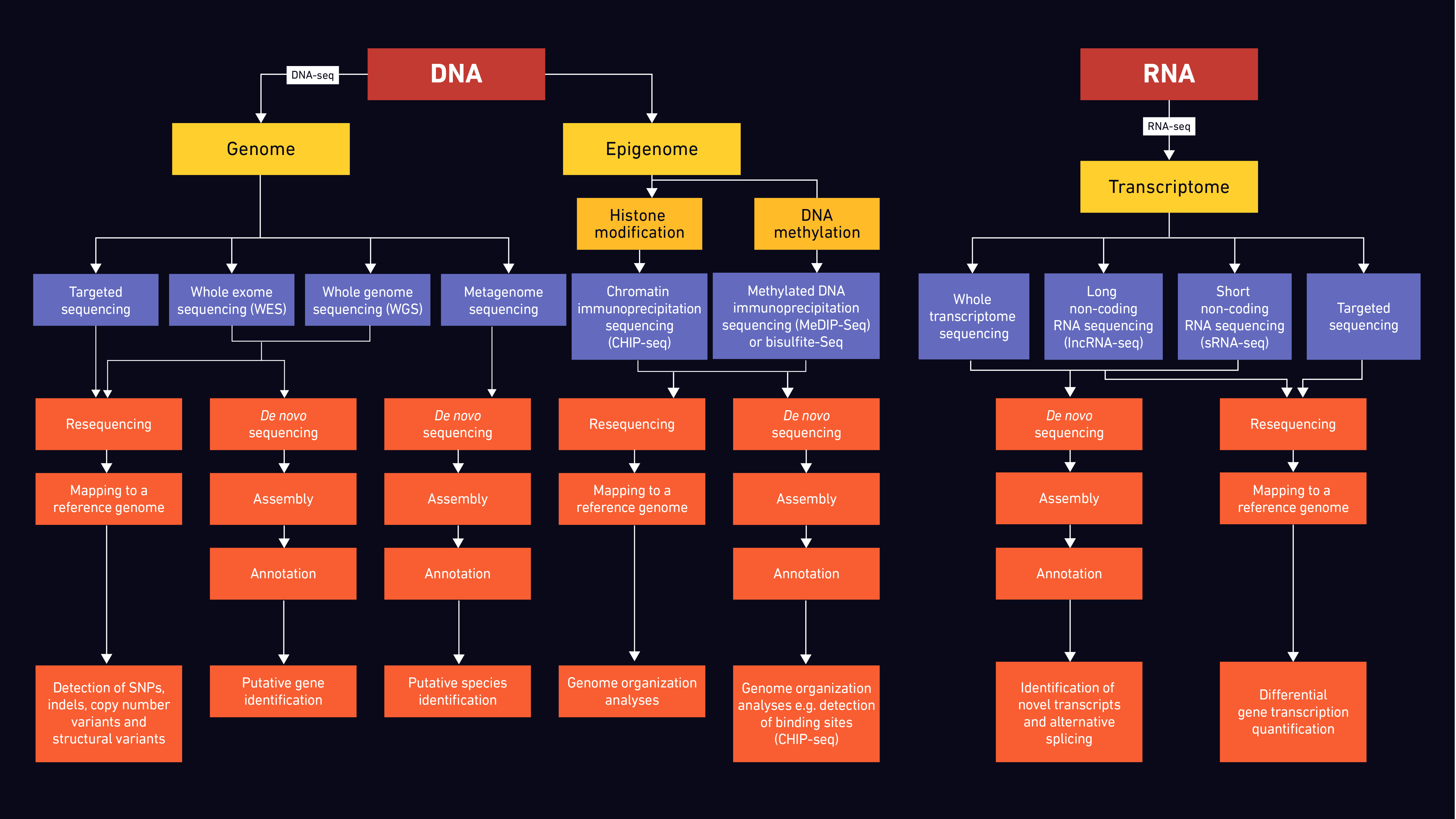
Generalised NGS Workflow
A standard NGS workflow for DNA and RNA typically follows the outline below.
Thank you for reading and hope you got to learn a thing or two regarding Next-Generation Sequencing.
Happy Learning.