




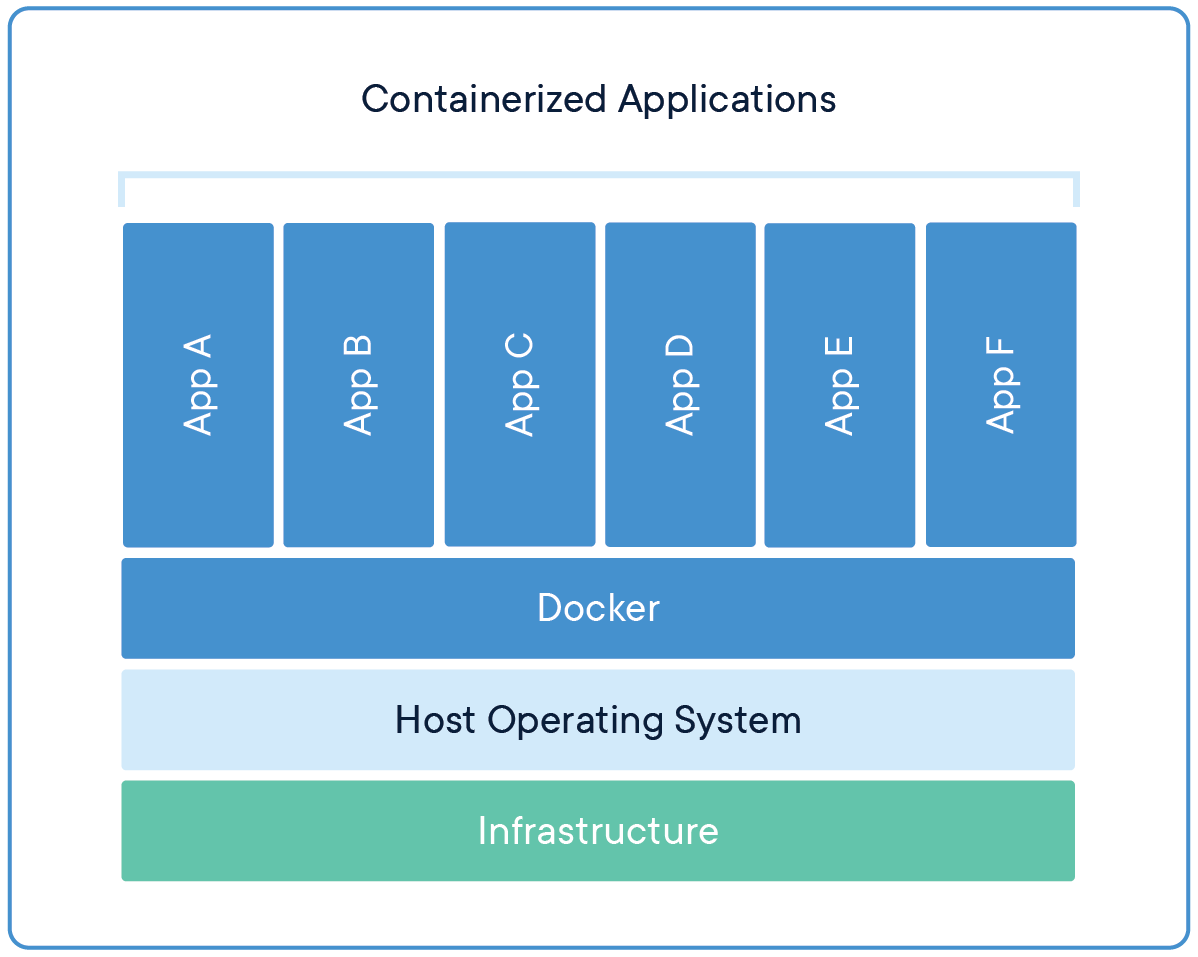
Containers are a technology for packaging and deploying software applications in a portable and isolated environment. They allow developers to bundle their application and its dependencies together in a single package, called a container. This package can then be run on any machine that has a container runtime, such as Docker or Singularity, installed.
Containers are different from traditional virtualisation, in that they do not create a fully virtualised operating system, but instead share the host operating system's kernel. This makes them more lightweight and faster to start than traditional virtual machines.
Containers offer several benefits for software development and deployment:
Reproducibility: Containers ensure that the application and its dependencies will run in the same way across different environments, making it easier to reproduce results and test new features.
Portability: Containers can be easily moved between different machines and environments, making it easier to deploy and scale applications.
Isolation: Containers provide a level of isolation between the application and the host system, which can help to improve security and prevent conflicts between different applications.
Versioning: Containers can be versioned, making it easy to roll back to a previous version of an application or revert to a specific configuration.
In the context of bioinformatics, containers are commonly used to package bioinformatics software and workflows, allowing researchers to run these tools in a consistent and reproducible environment, without worrying about dependencies and software compatibility.
Docker
Docker is a platform that allows developers and bioinformaticians to package and distribute their bioinformatics software and workflows in a consistent and reproducible way.
Docker uses containerization technology to create a portable and isolated environment for an application and its dependencies. This means that the software and its dependencies are bundled together in a container, which can be run on any machine with the Docker runtime installed. This eliminates the need for manual installation and configuration of dependencies and ensures that the software will run in the same way across different environments.
Docker also allows bioinformaticians to share their software and workflows with others in the community, by publishing their containers on the Docker Hub or other container registries. This enables other researchers to easily reproduce the results and use the software in their work.
In bioinformatics, Docker containers are commonly used to distribute software tools such as aligners, variant callers, RNA-seq and other bioinformatics pipelines that have complex dependencies. This allows researchers to run the software in a consistent and reproducible environment, without having to worry about software and dependencies compatibility.
Singularity
Singularity is an open-source containerization platform similar to Docker. It allows users to package their applications and dependencies together in a portable container, which can be run on any host machine with the Singularity runtime installed. Like Docker, Singularity enables the creation of reproducible and consistent environments for bioinformatics software and workflows.
However, Singularity was designed specifically for use in High-Performance Computing (HPC) and scientific computing environments where security, reproducibility, and performance are of paramount importance. Singularity allows for the use of existing system libraries and can be run on a cluster without the need for root access, which can be a concern in some HPC environments.
In summary, while Docker and Singularity share many similarities, Singularity was designed specifically for use in high-performance computing environments, providing extra features such as the ability to use existing system libraries, running on a cluster without the need for root access, which can be an important consideration for bioinformaticians working in HPC environments.
Pulling A Docker Container
To "pull" a Docker container means to download it from a container registry, such as Docker Hub, to your local machine. Once a container is pulled, it can be run using the docker run command.
The basic syntax for pulling a container is:
docker pull <image_name>
where <image_name> is the name of the container image you want to download.
For example, to pull the official Ubuntu 18.04 LTS image, you would use the command:
docker pull ubuntu:18.04
You can also specify a specific version of an image by adding the version tag after the image name, like this:
docker pull <image_name>:<tag>
For example, to pull version 1.0 of an image named "bioinformatics", you would use the command:
docker pull bioinformatics:1.0
You can also use a wildcard to pull all images that match a certain pattern. For example, to pull all images that are tagged as "bioinformatics" you can use the command :
docker pull bioinformatics*
Once you have pulled the image, you can run it by using the docker run command.
Keep in mind that pulling a container image can take some time, depending on the size of the image and your internet connection.
Creating A Dockerfile
A Dockerfile is a script that contains instructions for building a Docker container image. It specifies the base image to use, any additional software to install, and any configurations to make. Dockerfiles use a set of commands, called "instructions," to specify how to build a Docker image.
Here are some of the most commonly used instructions in a Dockerfile:
FROM: specifies the base image to use for the build.RUN: runs a command in the container during the build process.COPY: copies files from the host machine to the container.ENV: sets an environment variable in the container.EXPOSE: specifies the network ports that should be exposed by the container.CMD: specifies the command that should be run when the container is started.ENTRYPOINT: Specifies the command and its parameters that should be run when the container is started.USER: sets the UID (user ID) or username that the container should run as.WORKDIR: sets the working directory for subsequent instructions in the Dockerfile.VOLUME: creates a mount point for a volume in the container.LABEL: add metadata to the imageARG: defines a build-time variable, which can be passed at build-time to set the value of environment variables.
These are some of the most commonly used instructions in a Dockerfile, but there are many more commands and options available to customize your container as per your requirement.
Here is an example of a simple Dockerfile that installs the bioinformatics tool FastQC:
# Use the official Ubuntu image as the base
FROM ubuntu:20.04
# Update the package manager and install dependencies
RUN apt-get update && apt-get install -y \
wget \
unzip \
openjdk-11-jre
# Download and install FastQC
RUN wget <https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.9.zip> \
&& unzip fastqc_v0.11.9.zip \
&& rm fastqc_v0.11.9.zip \
&& chmod 755 FastQC/fastqc
# Add the FastQC executable to the PATH
ENV PATH="/FastQC:${PATH}"
# Set the working directory
WORKDIR /data
# Run the fastqc command when the container launches
CMD ["fastqc"]
The first line specifies the base image to use, in this case,
ubuntu:20.04The
RUNinstruction runs a command in the container. In this example, it updates the package manager and installs some dependencies (wget,unzipandopenjdk-11-jre)The next
RUNinstruction downloads and install FastQC by downloading the zip file from the website, unzipping it, removing the zip file and setting the permission of the folder.The
ENVinstruction sets environment variables for the container. In this case, it adds the path of the fastqc executable to the PATHThe
WORKDIRinstruction sets the working directory for any subsequent instructions.The
CMDinstruction specifies the command that should be run when the container starts.
Building Docker Image Using Dockerfile
Once you have created your Dockerfile, you can use the docker build command to build the container image. The basic syntax for building an image is:
docker build -t <image_name> .
Where -t flag is used to specify the name and optionally a tag to the name of the image in the name:tag format and . specifies the build context, that is the current directory.
For example, to build an image called "fastqc" from the Dockerfile in the current directory, you would use the command:
docker build -t fastqc .
Once the image is built, you can use the docker run command to start a container from the image and pass the input fastq files as arguments.
docker run -v /path/to/input_dir:/data fastqc sample1.fastq sample2.fastq
This command will run the fastqc command in the container, and create the output files in the input_dir on the host.
Converting Docker Image to Singularity
Singularity is a container platform that is designed to be more secure and performant than Docker in high-performance computing environments. To convert a Docker image to a Singularity image, you can use the singularity build command.
The basic syntax for converting a Docker image to a Singularity image is:
singularity build <image_name>.sif docker://<docker_image>:<tag>
Where <image_name>.sif is the name of the output Singularity image file, and docker://<docker_image>:<tag> is the name of the input Docker image.
For example, to convert a Docker image called "fastqc" to a Singularity image called "fastqc.sif", you would use the command:
singularity build fastqc.sif docker://fastqc:latest
Alternatively, you can use docker export or save command to export a container as a tarball and use sudo singularity import command to import the tarball into a singularity image
docker export <container_id> > container.tar
sudo singularity import <image_name>.sif container.tar
OR
docker save <image>:<tag> -o container.tar
singularity build <image>.sif docker-archive://container.tar
This will create a new singularity image called <image_name>.sif that you can use to start a container with the singularity run command.
Please note that while converting the image, it is important to check if the path and the environment variables are correctly set in the singularity image as it may vary from the original docker image and you might have to make adjustments accordingly.
Running A Docker|Singularity Container
Running a Docker container is done using the docker run command. The basic syntax for running a Docker container is:
docker run <image_name> <command>
Where <image_name> is the name of the Docker image, and <command> is the command to be executed inside the container.
For example, to run a Docker image called "fastqc" and execute the "fastqc" command, you would use the command:
docker run fastqc:<tag> fastqc <input_file>
Running a Singularity container is done using the singularity run command. The basic syntax for running a Singularity container is:
singularity run <image_name>.sif <command>
Where <image_name>.sif is the name of the Singularity image, and <command> is the command to be executed inside the container.
For example, to run a Singularity image called "fastqc.sif" and execute the "fastqc" command, you would use the command:
singularity run fastqc.sif fastqc <input_file>
You can also use the singularity shell command to open a shell session inside a Singularity container, similar to the docker run -it command for Docker. This allows you to interact with the container and execute multiple commands.
singularity shell <image_name>.sif
You can use exit command to exit from the shell session.
Please note that the singularity run command by default runs the container in the current working directory and with the same user permissions as the host machine. If you need to run the container in a different working directory or with different permissions, you can use the -B and -C options respectively.
Different Scenarios In Bioinformatics Tool Containerisation
Building an Ubuntu image using dockerfile
# Use a minimal version of Ubuntu as the base image FROM ubuntu:20.04 # Update package manager and install required dependencies RUN apt-get update && apt-get install -y wget # Set the command to execute on start CMD ['/bin/bash']Building Conda image with fastqc using dockerfile
# Use lighter miniconda3 to utilise conda FROM continuumio/miniconda3 # Update conda and install required packages to run FastQC RUN conda update -n base -c defaults conda && \ conda install -c bioconda fastqc # Set the command to execute on start CMD ["fastqc"]Building Conda image and activating the virtual environment
# Use lighter miniconda3 to utilise conda FROM continuumio/miniconda3:latest # Create a conda virtual env RUN conda create -n snpeff python=3.7 # Write the activate command to bashrc RUN echo "source activate snpeff" > ~/.bashrc # Set environment variable ENV PATH /opt/conda/envs/snpeff/bin:$PATH # Install required packages within the env RUN conda install -n snpeff -c conda-forge biopython pyvcf RUN conda install -n snpeff -c bioconda vcf-annotator # Set the command to execute on start CMD ['/bin/bash']Modifying packages/scripts within dockerfile
# Use lighter miniconda3 to utilise conda FROM continuumio/miniconda3:latest # Set environment variable ENV DISPLAY :0 # Update conda and set channels to search packages RUN conda update -n base -c defaults conda && \ conda config --add channels defaults && \ conda config --add channels bioconda && \ conda config --add channels conda-forge # Install qualimap package RUN conda install qualimap # To avoid: Creating plots Exception in thread "main" java.awt.AWTError: Can't connect to X11 window server using 'localhost:15.0' as the value of the DISPLAY variable # Using `sed` to replace java options from qualimap package RUN sed -i 's/java_options="-Xms32m -Xmx$JAVA_MEM_SIZE"/java_options="-Djava.awt.headless=true -Xms32m -Xmx$JAVA_MEM_SIZE"/g' /opt/conda/bin/qualimap # To avoid: Exception in thread "main" java.lang.Error: Probable fatal error:No fonts found RUN apt-get update -y && \ apt-get install -y fonts-liberation fonts-dejavu fontconfig xvfb # Set the command to execute on start CMD ["/bin/bash"]Multi-Staged builds
Multi-stage builds are a feature in Docker that allows you to use multiple
FROMinstructions in yourDockerfile. This allows you to use one image as a "builder" image to compile your code, and then use another image as the final "runtime" image, which only includes the necessary dependencies to run your application.The basic syntax for multi-stage builds is to use multiple
FROMinstructions in yourDockerfile, followed byCOPYinstructions to copy the necessary files from one stage to the next.#Stage 1: Build the miniconda3 environment stage1 FROM continuumio/miniconda3:latest as stage1 # Update conda and install fastqc RUN conda update -n base -c defaults conda && \ conda create -n fastqc python=3.7 && \ echo "source activate fastqc" > ~/.bashrc # Set environment variable ENV PATH /opt/conda/envs/fastqc:$PATH # Install fastqc RUN conda install -n fastqc -c bioconda fastqc #Stage 2: Build the GATK layer FROM broadinstitute/gatk:latest # Copy contents of conda installation from previous stage1 build COPY --from=stage1 /opt/conda /opt/conda # Write the activate command to bashrc RUN echo "source activate fastq" > ~/.bashrc # Set environment variable ENV PATH /opt/conda/envs/fastq/bin:$PATH # Set the command to execute on start CMD ["/bin/bash"]Multi-stage builds allow you to keep the final image minimal by only including the necessary dependencies for runtime and not including build dependencies or any unnecessary files. This helps to reduce the image size, improve security, and increase performance.
It is important to note that the intermediate build stages are not stored in the image repository and you can only reference the last stage of the build.
Thank you for reading and hope you got to learn a thing or two regarding containers.
Happy Learning.