




Table of contents
- Introduction
- Why QC Of NGS Data Is Important?
- Steps In QC Of NGS Data
- Difference In QC Steps For WES, WGS and RNAseq Data
- Difference In QC Steps For Short and Long Read Sequencing
- Parameters To Consider While Choosing a Sequence Cleaning tool
- Tools utilized for cleaning fastq sequences
- What are FastQC and MultiQC?
- Using QC Tools
Introduction
Next-generation sequencing (NGS) technologies have revolutionized the field of genomics by enabling researchers to generate large amounts of sequence data quickly and at a lower cost than traditional sequencing methods. However, raw NGS data can be prone to errors and biases, which can affect downstream analysis and interpretation.
Sequence cleaning, also known as data preprocessing or quality control, is a crucial step in NGS data analysis that involves removing low-quality reads, adapter sequences, and potential contaminants from the raw sequencing data.
In this article, we will explore the importance of sequence cleaning in NGS data analysis, review some of the commonly used sequence cleaning tools, and discuss key parameters to consider when choosing a sequence cleaning tool for your data.
Why QC Of NGS Data Is Important?
Quality control (QC) of next-generation sequencing (NGS) data is a crucial step in the data analysis pipeline. Here are some of the reasons why QC is important:
Identify errors and biases: NGS data can be prone to errors and biases due to a variety of factors such as sample quality, library preparation, and sequencing technology. QC helps to identify and correct these errors and biases, which can affect downstream analysis and interpretation.
Improve data quality: Removing low-quality reads and potential contaminants can improve the overall quality of the sequencing data, which can lead to more accurate and reliable results.
Save time and resources: QC can help to identify problems early in the analysis pipeline, which can save time and resources by preventing the analysis of low-quality or contaminated data.
Compare data across samples: QC metrics can be used to compare data across different samples or experiments, allowing for more meaningful comparisons and insights.
Ensure reproducibility: Consistent QC practices can help to ensure the reproducibility of the data and analysis pipeline, which is important for scientific rigour and validity.
In summary, QC of NGS data is a critical step in the analysis pipeline that helps to improve data quality, identify errors and biases, save time and resources, and ensure reproducibility.
Steps In QC Of NGS Data
Adapter and primer trimming
Adapter and primer trimming is a quality control step in NGS data analysis that involves removing sequences corresponding to adapter and primer sequences that were added during library preparation. These adapter and primer sequences can interfere with downstream analysis and affect the accuracy of read alignment and variant calling.
Adapters are short, synthetic DNA sequences that are ligated to the ends of DNA fragments during library preparation to facilitate sequencing. Primers are short, synthetic DNA sequences that are used to amplify specific regions of interest, such as in PCR-based library preparation methods. During sequencing, adapters and primers can become incorporated into the resulting sequencing reads and cause issues such as reduced mapping efficiency, false positive variant calls, and other downstream analysis errors.
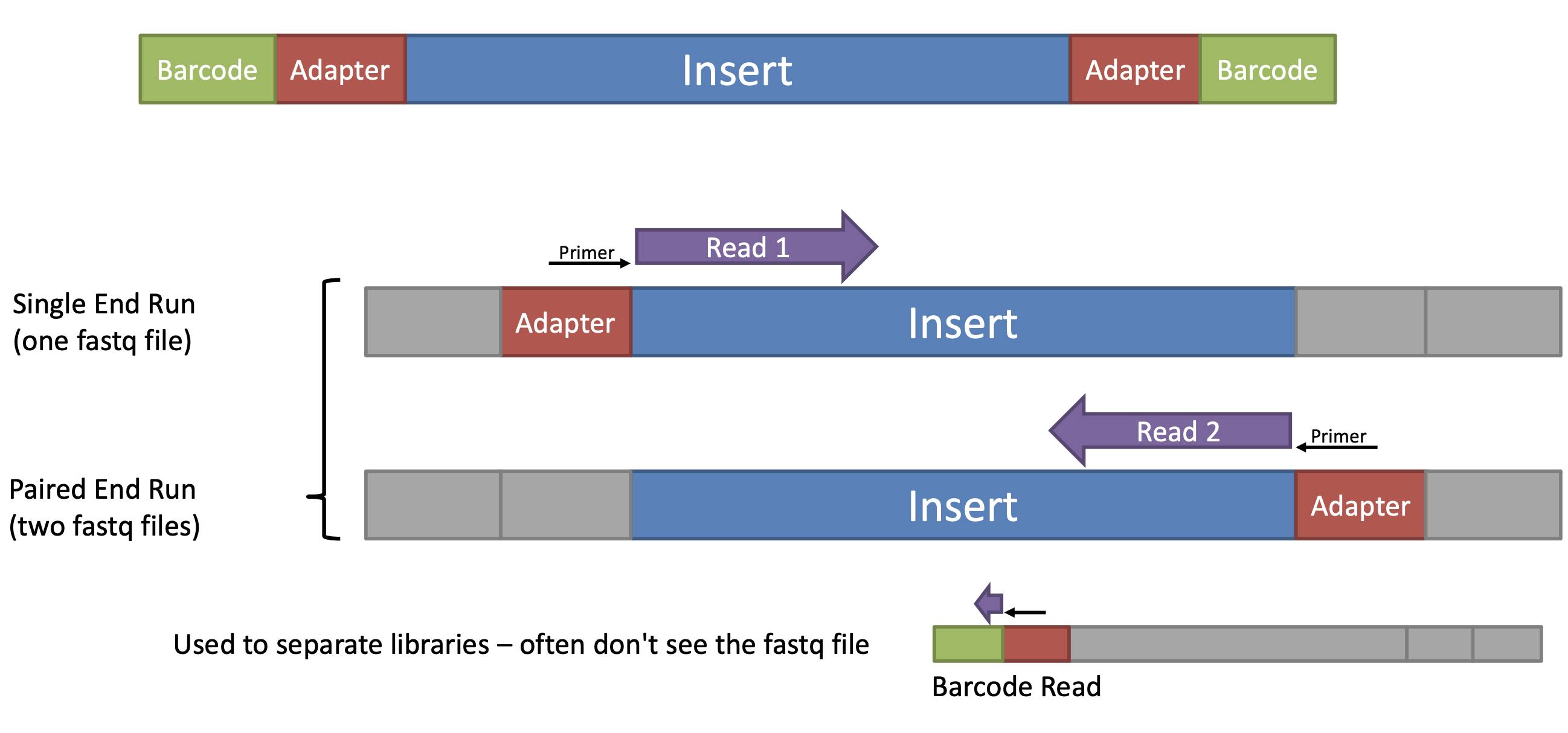
Adapter and primer trimming software tools, such as Trimmomatic, Cutadapt, and BBDuk, can identify and remove these sequences from the raw sequencing data, leaving behind only the high-quality reads that can be used for downstream analysis. The trimming process typically involves aligning the reads to a reference database of adapter and primer sequences, identifying the adapter and primer sequences in the reads, and trimming them off before further analysis.
Adapter and primer trimming is an important QC step in NGS data analysis, as it can help to improve the quality and accuracy of the sequencing data and reduce the potential for errors and biases in downstream analysis.
Others
In addition to adapter and primer trimming, there are several other important quality control (QC) steps that are typically performed on fastq files in next-generation sequencing (NGS) data analysis. These include:
Quality score filtering: Fastq files contain quality scores that indicate the confidence level of each base call. Quality score filtering removes low-quality reads or bases based on a minimum quality score threshold. This helps to ensure that the data used in the downstream analysis is of high quality and reduces the potential for errors and biases.
Read length filtering: Reads that are too short or too long can be removed based on a specified length threshold. This can help to improve the accuracy of downstream analysis and reduce computational resources needed for analysis.
Duplicate removal: PCR duplicates can artificially inflate read counts and lead to an overestimation of true biological diversity. Duplicate removal helps to reduce this effect and improve the accuracy of downstream analysis.
Contaminant removal: Fastq files can contain reads that originate from sources other than the sample of interest, such as environmental contaminants or sequencing artefacts. Contaminant removal helps to identify and remove these reads from the data to improve the accuracy of downstream analysis.
Quality control metric calculation: Several QC metrics, such as the number of reads, read length distribution, and GC content, can be calculated to assess the quality and suitability of the data for downstream analysis.
These QC steps are important for ensuring the accuracy and reliability of downstream analysis and interpretation of NGS data.
By performing thorough QC on fastq files, researchers can identify and correct errors and biases in the data, reduce the potential for false positives and false negatives, and increase the overall quality and usefulness of the data.
Difference In QC Steps For WES, WGS and RNAseq Data
The quality control (QC) steps for sequencing data may differ slightly depending on the type of sequencing data being analyzed. Here are some general differences in the QC steps for whole exome, whole genome, and RNAseq data:
Whole Exome Sequencing (WES):
Coverage analysis is crucial to ensure adequate depth of coverage for all regions of the exome.
The distribution of read depths across the exome should be examined to identify any regions with low coverage, which may require additional sequencing or targeted enrichment.
The variant calling step should be validated using a reference sample with known genotypes or using other tools such as Sanger sequencing.
Whole Genome Sequencing (WGS):
Similar to WES, coverage analysis is important to ensure adequate depth of coverage across the entire genome.
The distribution of read depths should be examined to identify regions with low coverage, which may require additional sequencing or targeted enrichment.
Quality control measures such as base quality score recalibration, duplicate read removal, and variant quality score recalibration should be performed to improve the accuracy of variant calling.
RNAseq:
Quality control of RNAseq data typically includes examining basic statistics such as read count, mapping rate, and gene coverage.
Transcript quantification and differential expression analysis should be validated using tools such as qPCR or alternative splicing analysis.
Additional steps may be required to account for biases introduced during library preparation and sequencing, such as GC bias correction and rRNA depletion.
In general, QC for any type of sequencing data should include measures to assess read quality, such as per-base and per-sequence quality scores, and identify and remove any potential contaminants. However, the specific steps and metrics used may vary depending on the type of data and the specific research question being addressed.
Difference In QC Steps For Short and Long Read Sequencing
The QC step can differ between short-read and long-read sequencing data due to several factors, such as the characteristics of the sequencing technology, the type of library preparation, and the type of downstream analysis.
Here are some differences in QC steps between short-read and long-read sequencing data:
Adapter and primer trimming: Similar to short read data, adapter and primer trimming is typically performed on long read sequencing data to remove adapter and primer sequences that can interfere with downstream analysis. However, since long-read sequencing technologies such as PacBio and Oxford Nanopore use different library preparation methods, specific adapter and primer trimming tools may need to be used for long-read data.
Quality score filtering: Quality score filtering is important for both short and long-read data to ensure that low-quality reads or bases are removed from the data. However, the quality score distribution and the methods used to calculate quality scores can differ between short and long-read data, and therefore may require different quality score filtering parameters or algorithms.
Read length filtering: Read length filtering is typically more important for short read data since short reads are typically generated with a fixed length and reads that are too short or too long can be removed to improve downstream analysis. Long-read sequencing technologies can generate reads that vary greatly in length, so read-length filtering may be less critical for long-read data.
Duplicate removal: Duplicate removal is important for both short and long-read data, but the methods used to identify and remove duplicates can differ between the two types of data. For short-read data, PCR duplicates can be identified based on identical read sequences, while for long-read data, duplicates can be identified based on the location and orientation of the reads in the genome or transcriptome.
Base modification and error correction: Long-read sequencing technologies can generate reads that contain base modifications, such as DNA methylation, which can affect downstream analysis. Long-read QC can include steps to identify and correct these base modifications to improve the accuracy of downstream analysis.
In summary, while some QC steps are similar between short-read and long-read sequencing data, such as adapter and primer trimming and duplicate removal, differences in the characteristics of the data can require different QC approaches and tools. It is important to carefully consider the specific characteristics of the sequencing data and the downstream analysis when designing a QC pipeline for short-read or long-read data.
Here is a table comparing the QC steps between short-read and long-read sequencing:
| QC Step | Short Read Sequencing | Long Read Sequencing |
| Adapter and primer trimming | Performed using specific tools such as Cutadapt or Trimmomatic | Performed using specific tools such as Porechop or Nanocut |
| Quality score filtering | Thresholds based on quality score distributions | Thresholds based on quality score distributions |
| Read length filtering | Typically performed to remove reads that are too short or too long | Maybe less critical due to variable read lengths |
| Duplicate removal | Identical sequences are flagged as PCR duplicates | Duplicates are identified based on location and orientation |
| Base modification and error correction | Base modifications such as DNA methylation can be corrected using specific tools | Base modifications such as DNA methylation can be corrected using specific tools |
It is important to note that the specific QC steps and tools used can vary depending on the sequencing technology and library preparation method.
Parameters To Consider While Choosing a Sequence Cleaning tool
When choosing a sequence cleaning tool, several parameters should be considered, including:
Type of sequencing data: Some tools are specifically designed for certain types of sequencing data, such as single-end or paired-end reads. It's important to choose a tool that can handle the specific data type.
Quality of data: Different tools have different approaches to quality filtering and trimming. Some tools may be more aggressive in removing low-quality bases, while others may be more conservative. Consider the quality of the data and the desired level of filtering when choosing a tool.
Adapter sequences: Some tools are better than others at detecting and removing adapter sequences. Consider the type of adapter used in the sequencing library and whether the chosen tool can handle it.
Computational resources: Some tools may require more computational resources than others, particularly for large datasets. Consider the available resources, such as RAM and processing power, when choosing a tool.
Customizability: Some tools are highly customizable, allowing users to adjust parameters and settings to suit their specific needs. Consider the level of customization required for the specific project when choosing a tool.
Compatibility: Some tools may be more compatible with certain operating systems or programming languages. Consider the compatibility with the existing software infrastructure when choosing a tool.
Tools utilized for cleaning fastq sequences
Adapter and Primer Trimming tools
Trimmomatic - Trimmomatic is a widely used tool for removing adapter sequences, trimming low-quality bases, and removing reads below a certain length threshold. It can be used to clean both single-end and paired-end reads.
Cutadapt - Cutadapt is a tool specifically designed for adapter trimming. It is fast and flexible, allowing for the removal of adapters with different lengths and mismatches. It can also perform quality trimming and filtering of short reads.
BBDuk - BBDuk is a tool that can be used for adapter trimming, quality filtering, and contaminant removal. It is highly customizable and can handle both single-end and paired-end reads.
AdapterRemoval - AdapterRemoval is a tool for removing adapter sequences and trimming low-quality bases from sequencing data. It can also merge overlapping paired-end reads and can handle data with different read lengths.
Skewer - Skewer is a tool for adapter trimming and quality filtering of sequencing data. It uses a dynamic programming algorithm to align reads to adapter sequences and can handle both single-end and paired-end reads.
SeqPurge - SeqPurge is a tool for adapter and quality trimming of sequencing data. It uses a probabilistic model to identify and remove adapters and can handle both single-end and paired-end reads.
Cutadapt-ng - Cutadapt-ng is a fork of the original Cutadapt tool that has been optimized for speed and memory usage. It can perform adapter trimming and quality filtering of sequencing data and can handle both single-end and paired-end reads.
rRNA Filtering Tools
Ribodetector - Ribodetector is a tool for detecting rRNA contamination in RNA-seq data. It uses a database of rRNA sequences to align reads and identify rRNA reads. It can also remove rRNA reads from the data set.
SortMeRNA - SortMeRNA is a tool that can be used for filtering rRNA sequences from sequencing data. It uses a reference database of rRNA sequences to align reads and identify rRNA reads. It can handle both single-end and paired-end reads and can be used to filter out other types of RNA as well.
Decontam - Decontam is a tool for identifying and removing contaminants from sequencing data, including rRNA sequences. It uses statistical models to identify contaminant reads based on their frequency and other characteristics and can be used to filter out both host and environmental contaminants.
Contamination Detection Tools
KneadData - KneadData is a tool for identifying and removing contaminating reads from sequencing data. It uses a reference database of genomes and can be used to filter out reads from host organisms, bacterial contaminants, and other types of contaminants. It can also perform quality trimming and adapter removal.
BlobTools - BlobTools is a tool for taxonomic profiling and contamination detection of sequencing data. It uses a reference database of genomes to classify reads at different taxonomic levels and can be used to identify contaminating reads from host organisms or other sources.
KrakenUniq - KrakenUniq is a tool for the taxonomic classification of sequencing data that can be used to identify and remove duplicate reads. It uses a reference database of genomes and can classify reads at different taxonomic levels. Removing duplicate reads can reduce the amount of computational resources required for downstream processing.
ConDeTri - ConDeTri is a tool for detecting and removing contamination from sequencing data. It uses a statistical model to identify contaminating reads based on their frequency and other characteristics and can be used to filter out reads from host organisms, bacterial contaminants, and other sources.
These tools can be particularly useful for identifying and removing contaminants from sequencing data, which can improve the accuracy and quality of downstream analyses. By filtering out contaminating reads, researchers can reduce the number of errors introduced during sequencing and increase the sensitivity of their analyses. Overall, these tools should be part of any fastq sequence cleaning pipeline.
Resources For Cleaning Long Read Sequencing Data.
Porechop - Porechop is a tool that can be used for adapter trimming and demultiplexing of Oxford Nanopore reads. It uses a dynamic programming algorithm to align reads to adapter sequences and can also demultiplex reads based on barcode sequences.
Filtlong - Filtlong is a tool for filtering long reads based on length and quality. It can be used to remove low-quality reads, as well as reads that are too short or too long for downstream analyses.
Nanofilt - Nanofilt is a tool for filtering long reads based on quality, length, and other criteria. It can be used to remove low-quality reads, adapter sequences, and reads that are too short or too long.
MiniBar - MiniBar is a tool for removing adapter sequences and filtering low-quality regions from PacBio long reads. It uses a seed-and-extend algorithm to align reads to adapter sequences and can also filter out low-quality regions using a sliding window approach.
These tools are essential for cleaning long-read sequencing data, which can have higher error rates and more complex error profiles than short-read data. By filtering out low-quality reads and adapter sequences, researchers can improve the accuracy and quality of downstream analyses.
The following table provides a more concise overview of the various tools that can be used for cleaning and filtering sequencing data, please note this is not a comprehensive list.
| Tool Name (Date) | Type of Sequencing | Input | Output | Function |
| Trimmomatic (2011) | Short | FASTQ files | Trimmed FASTQ files | Trimming adapters and low-quality reads |
| Cutadapt (2011) | Short | FASTQ files | Trimmed FASTQ files | Adapter trimming and quality filtering |
| Ribodetector (2011) | Short/Long | FASTQ files | FASTA/Q files without rRNA reads | rRNA filtering |
| ContaminantFilter (2015) | Short/Long | FASTQ files | FASTQ files without contaminants | Contaminant filtering |
| SortMeRNA (2016) | Short/Long | FASTQ files | FASTQ files without rRNA reads | rRNA filtering |
| Decontam (2018) | Short/Long | FASTQ files | FASTQ files without contaminants | Contaminant filtering |
| KneadData (2019) | Short/Long | FASTQ files | FASTQ files without contaminants | Contaminant filtering |
| BlobTools (2019) | Short/Long | FASTQ files, assembly, annotation | Taxonomic and contamination reports | Taxonomic profiling and contamination detection |
| KrakenUniq (2019) | Short/Long | FASTQ files | Taxonomic classification report | Taxonomic classification and duplicate removal |
| ConDeTri (2020) | Short/Long | FASTQ files | FASTQ files without contaminants | Contaminant filtering |
| Porechop (2017) | Long | Fast5/Fastq files | Adapter-trimmed Fastq files | Adapter trimming and demultiplexing for Nanopore reads |
| Filtlong (2018) | Long | Fastq files | Filtered Fastq files | Quality filtering for long reads |
| Nanofilt (2019) | Long | Fastq files | Filtered Fastq files | Quality and adapter filtering for long reads |
| MiniBar (2020) | Long | Fastq files | Filtered Fastq files | Adapter trimming and low-quality filtering for PacBio reads |
What are FastQC and MultiQC?
FastQC is a software tool that performs quality control checks on high-throughput sequencing data generated from both short and long reads. It generates a report that summarizes various metrics such as base quality, sequence length distribution, GC content, adapter content, overrepresented sequences, and per-base sequence content.
These metrics can help identify problems in the data that may arise from sequencing or sample preparation issues, allowing the user to make informed decisions about downstream data processing.
MultiQC, on the other hand, is a software tool that generates a single report by aggregating and summarizing the results from multiple FastQC runs. This is particularly useful when working with large datasets, as it allows the user to quickly and easily compare the quality of multiple samples in a single report.
The report generated by MultiQC includes the same metrics as FastQC, with the addition of an overview of the data and a summary of the most common issues across all samples.

Following are examples of MultiQC reports for a different combination of tools for each analysis type:
WGS Report: https://multiqc.info/examples/wgs/multiqc_report.html
RNASeq Report: https://multiqc.info/examples/rna-seq/multiqc_report.html
Using QC Tools
Docker and Singularity are containerization tools that allow users to package software applications and their dependencies into portable containers, making it easy to deploy and run the software across different computing environments. Using these tools can be particularly useful when working with quality control (QC) tools for sequencing data, as they often require specific software versions and configurations to run properly.
By using Docker or Singularity containers, users can ensure that the QC tools are running in a consistent environment, with all necessary dependencies already installed. This eliminates the need for users to manually install and configure the software on their local machines, which can be time-consuming and error-prone. Additionally, containers can be easily shared and deployed across different computing environments, making it easier for researchers to collaborate and reproduce analyses. Overall, using Docker or Singularity containers can simplify the process of using QC tools for sequencing data and help ensure reproducible research practices.
Fastp is a popular tool for quality control and adapter/primers trimming of high-throughput sequencing data. In addition to its ability to remove adapters and low-quality bases, Fastp has a built-in algorithm for identifying and trimming PCR primers from sequencing reads.
This can be particularly useful for amplicon sequencing studies or other sequencing approaches that utilize PCR amplification. To use fastp for primer trimming, users can specify the primer sequences using the -p or --detect_adapter_for_pe options, depending on whether the sequencing data is paired-end or single-end.
Here's an example command for using Fastp to trim primers from paired-end sequencing data:
fastp -i input_R1.fastq.gz -I input_R2.fastq.gz \
-o output_R1.fastq.gz -O output_R2.fastq.gz \
--detect_adapter_for_pe -p primer_seq1 -p primer_seq2
In this command, input_R1.fastq.gz and input_R2.fastq.gz are the input paired-end sequencing data files, and output_R1.fastq.gz and output_R2.fastq.gz are the trimmed output files. The --detect_adapter_for_pe option tells fastp to automatically detect and trim adapters from the sequencing data, and the -p option specifies the primer sequences to be trimmed. Users can specify multiple primer sequences using multiple -p options.
To use fastp within a Docker container, users can first pull the fastp container from Docker Hub using the following command:
docker pull staphb/fastp:latest
Once the container is downloaded, users can run fastp within the container using the following command:
docker run --rm -v /path/to/local/files:/data staphb/fastp fastp \
-i /data/input_R1.fastq.gz -I /data/input_R2.fastq.gz \
-o /data/output_R1.fastq.gz -O /data/output_R2.fastq.gz \
--detect_adapter_for_pe -p primer_seq1 -p primer_seq2
In this command, the -v option is used to mount the local input and output files to the container, and the staphb/fastp:latest argument specifies the name and version of the Fastp container. The Fastp command and its options are specified after the container name.
One benefit of using a Docker container to run Fastp is that it ensures that the tool is running in a consistent environment with all necessary dependencies already installed. This eliminates the need for users to manually install and configure the tool on their local machines, which can be time-consuming and error-prone.
Additionally, containers can be easily shared and deployed across different computing environments, making it easier for researchers to collaborate and reproduce analyses. Overall, using Docker containers can simplify the process of running Fastp and help ensure reproducible research practices.
Thank you for reading and I hope you got to learn a thing or two regarding sequence cleaning of NGS data.
To gain a general understanding of next-generation sequencing (NGS) or to refresh your knowledge on the topic, please refer to the following article.
Happy Learning.