


NGS Data Analysis: Nextflow DSL2
Nextflow - Because Life's Too Short for Bash Scripts!


Introduction
Next Generation Sequencing (NGS) has revolutionized genomics research, enabling scientists to decode vast amounts of genetic information rapidly. However, the power of NGS comes with the challenge of efficiently handling and analyzing the colossal datasets it produces.
In the world of genomics, effective data analysis is as crucial as data generation itself. This article introduces Nextflow, a game-changing tool for NGS data analysis, and dives into the creation of a Sequence Quality Control (QC) pipeline using Nextflow.
A. Overview of NGS Data Analysis
NGS generates a myriad of data formats, ranging from raw sequence reads (FASTQ) to processed variant calls (VCF). This diversity brings about significant analytical challenges, including managing data volume, dealing with data complexity, and ensuring the reproducibility of analyses.
To tackle these challenges we can opt for workflow management systems like Nextflow. Read more about the overview of NGS and the steps in the following article:
Nextflow's DSL2 (Domain-Specific Language 2) is the second generation of Nextflow DSL, which is a powerful and flexible scripting language designed specifically for defining and managing data analysis workflows. DSL2 builds upon the features of the original DSL and introduces enhancements to make workflow creation even more intuitive and expressive.
B. The Importance of Efficient Data Analysis
Efficient data analysis is paramount in NGS experiments. It not only saves researchers valuable time but also ensures that the results are reliable and reproducible.
Without a robust analysis framework, researchers may struggle to handle the intricacies of NGS data, leading to errors and inefficiencies that can hinder reproducibility in scientific research.
Alrighty, folks, let's dive into the nitty-gritty of Nextflow, the tool that's about to make your NGS data analysis life a whole lot easier. We all know that dealing with Next Generation Sequencing (NGS) data can be like herding cats, but fear not, because Nextflow is here to save the day.
In this article, we're going to break down some key elements of a Nextflow workflow script for a project we'll call 'SequenceCleaning.' Picture this: you've got a bunch of messy sequencing data (those FASTQ files) that need some serious quality control TLC. We'll show you how Nextflow, along with a sprinkle of Docker magic, can help you whip your data into shape.
So, grab your favorite beverage, put on your coding hat, and let's explore how to set up the SequenceCleaning project using Nextflow.
Introduction to Nextflow
Nextflow is an innovative workflow management system designed explicitly for scientific data analysis. It simplifies the creation, execution, and management of complex NGS workflows. By leveraging the power of Nextflow, researchers can streamline their NGS data analysis pipelines, making them more reproducible, scalable, and user-friendly.
Advantages of Using Nextflow over Bash Scripts
Reproducibility: Nextflow allows you to specify software and environment dependencies, ensuring that your analysis remains reproducible across different computing environments. In contrast, Bash scripts may lack this level of version control and dependency management.
Parallelization: Nextflow excels in distributing tasks across multiple CPUs or even clusters effortlessly. Bash scripts may require manual efforts to achieve similar parallelism, making Nextflow a more efficient choice for large-scale analyses.
Error Handling: Nextflow provides robust error handling and reporting mechanisms, which simplify debugging and troubleshooting. In Bash scripts, error handling often involves custom solutions, making the process more error-prone.
Portability: Nextflow workflows can be easily shared and reused across different research groups, as they encapsulate all necessary dependencies and settings. Bash scripts may rely on specific system configurations, making them less portable.
Community and Support: Nextflow has an active and supportive user community, with readily available documentation and resources. Bash scripts may lack the same level of community support and standardized best practices.
In the world of NGS data analysis, Nextflow stands as a powerful ally, simplifying the complex journey from raw sequence data to meaningful insights.
In the following sections, we will delve deeper into Nextflow, exploring its capabilities through a practical Sequence QC pipeline example.
A. Files Setup
To set up the file structure for a Nextflow project called "SequenceCleaning," where "SequenceCleaning" is the parent folder with subfolders for modules and containing configuration and workflow files, you can follow this directory structure:
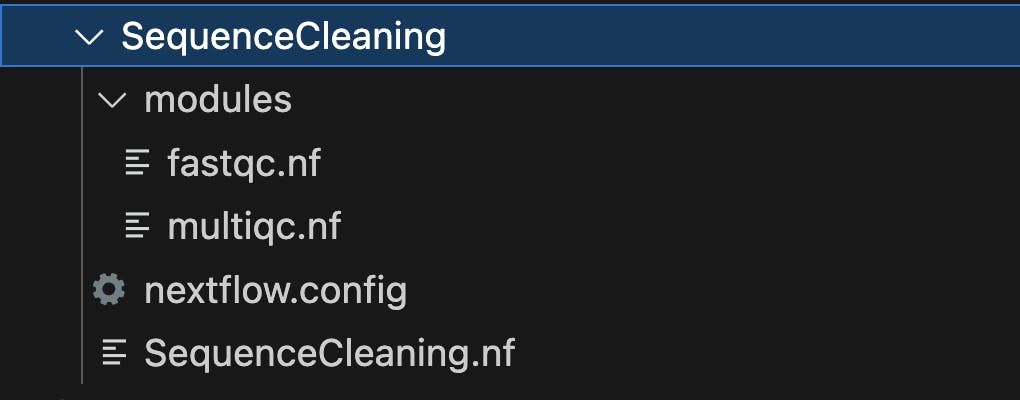
Let's briefly explain the purpose of each of these files and folders:
SequenceCleaning/: This is the main project directory, which serves as the parent folder for your Nextflow project. It contains all the necessary files and subdirectories for organizing your workflow.nextflow.config: This file is where you specify configuration settings for your Nextflow project. It allows you to define parameters such as the executor (e.g., local, cluster, cloud), resource allocation, and any custom settings required for your workflow.SequenceCleaning.nf: This is the primary Nextflow workflow script for your Sequence Cleaning project. Inside this script, you define the structure of your workflow, including the processes, inputs, outputs, and dependencies. This is where you'll incorporate and reference the modules defined in the "modules" subfolder.modules/: This is a subdirectory where you can organize and store individual workflow modules. Modules are reusable components of your workflow that encapsulate specific tasks or processes. In your case, you have two modules,fastqc.nfandmultiqc.nfwhich contain the instructions for running FastQC and MultiQC, respectively.
Here's a brief overview of how this file structure works in a Nextflow project:
The
nextflow.configfile contains project-wide configuration settings.The main workflow logic is defined in
SequenceCleaning.nf, which references the modules within the "modules" subfolder.The "modules" subfolder houses the individual process definitions for specific tasks, allowing for modularity and reusability of workflow components.
With this setup, you can easily manage and expand your Sequence Cleaning project using Nextflow, taking advantage of modularity and configurability to streamline your NGS data analysis.
B. Nextflow Config
params {
reads = "/completepath/data/*_{1,2}.fastq.gz"
output = "$baseDir/output"
}
docker {
enabled = true
containerOptions = '--rm -u $(id -u):$(id -g)'
}
process {
cpu = 2
memory = 2.GB
withName: FASTQC {
container = 'staphb/fastqc:latest'
}
withName: MULTIQC {
container = 'staphb/multiqc:latest'
}
}
This is a configuration file for our Nextflow workflow, and it sets various parameters and options for running the workflow. Let's break down what each section of the code does:
paramsBlock:reads: This parameter specifies the input data for the workflow. It uses a glob pattern to match FASTQ files (ending in ".fastq.gz") in a directory located at "/completepath/data/." The pattern "{1,2}" indicates that it's looking for files with "_1" or "_2" in their names, which is common in paired-end sequencing data.output: This parameter specifies the output directory where the results of the workflow will be stored. The$baseDirvariable represents the base directory of the workflow, and it appends "/output" to that path.
dockerBlock:enabled = true: This setting enables the use of Docker containers for running the processes in the workflow. Docker is a platform for developing, shipping, and running applications in containers, which can provide a consistent and isolated environment for your workflow processes.containerOptions: This option specifies additional options to be passed to the Docker container when it's executed. It uses the--rmoption to automatically remove containers after they finish running, and it sets the user ID (-u) and group ID (-g) inside the container to match the user's ID and group ID running the Nextflow workflow. This helps ensure that the files created inside the container are owned by the user running the workflow.
processBlock:cpu = 2: This setting specifies that each process in the workflow should be allocated 2 CPU cores.memory =2.GB: This setting specifies that each process should be allocated 2 gigabytes of memory (RAM).
The withName blocks specify additional settings for processes with specific names:
withName: FASTQC: This block specifies that processes with the name "FASTQC" should run in a Docker container with the image "staphb/fastqc:latest." This means that the "FASTQC" process will use the specified Docker container to execute its tasks.withName: MULTIQC: Similarly, processes with the name "MULTIQC" will run in a Docker container with the image "staphb/multiqc:latest."
In summary, this code block sets various parameters and options for a Nextflow workflow. It defines input data patterns, output directories, enables Docker containerization for processes, and configures resource allocation for CPU and memory. Additionally, it specifies Docker container images for processes with specific names, allowing for the isolation and reproducibility of the analysis steps in the workflow.
Consider the provided configuration above as your trusty blueprint whenever you embark on the exciting journey of crafting your own pipeline.
If you ever find yourself pondering the mysteries of Docker or need guidance on how to harness its powers, be sure to check out the following article for all the answers:
C. Nextflow modules
Owning a Mac can have its perks, for instance, installing tools like Fastp for adapter trimming or getting Docker containers to cooperate can be a bit of a challenge, especially with the new silicon architecture. We're keeping our fingers crossed, hoping that some kind-hearted developers will come to the rescue. In the meantime, let's roll with FastQC and MultiQC.
PS. one can use the same code structure as FastQC module nextflow script for fastp or any (adapter trimming) tool.
- FastQC Module
process FASTQC {
tag "${sample_id}"
publishDir "${params.output}", mode: 'copy'
input:
tuple val(sample_id), path(reads)
output:
path("${sample_id}_fastqc_logs/*.{zip,html}")
script:
"""
mkdir -p ${sample_id}_fastqc_logs
fastqc -o ${sample_id}_fastqc_logs \
-f fastq \
-q ${reads} \
-t ${task.cpus}
"""
}
The provided code defines a Nextflow module called fastqc.nf that encapsulates the process of running FastQC, a popular quality control tool for Next Generation Sequencing (NGS) data. Let's break down the code to understand its components:
Process Definition (FASTQC):
- This module defines a process named "FASTQC" using the
processkeyword. In Nextflow, a process represents a computational task or step in the workflow.
- This module defines a process named "FASTQC" using the
tag "${sample_id}":- The
tagdirective assigns a unique tag to each execution of the process. In this case, it uses the${sample_id}variable as the tag. This helps in organizing and identifying individual process runs when the workflow is executed.
- The
publishDir "${params.output}", mode: 'copy':- The
publishDirdirective specifies where the output files generated by this process should be published or copied. It uses the${params.output}parameter, which typically points to the output directory defined in the workflow's configuration. This ensures that FastQC output files will be collected and organized in the specified output directory.
- The
Input Section:
The
inputsection defines the inputs required for this process:tuple val(sample_id), path(reads): This input specification expects a tuple consisting ofsample_id(a unique identifier for the sample being processed) and thereadsinput path, which represents the path to the input FASTQ file(s).
Output Section:
The
outputsection defines the expected output of this process:path("${sample_id}_fastqc_logs/*.{zip,html}"): This output specification defines a directory structure where FastQC output files in ZIP and HTML formats will be saved. The${sample_id}variable is used to create a unique directory for each sample.
Script Section:
The
scriptsection contains the actual shell script that will be executed by this process. It performs the following actions:Creates a directory with the name
${sample_id}_fastqc_logsto store FastQC output files.Executes the FastQC tool with the following parameters:
-o ${sample_id}_fastqc_logs: Specifies the output directory for FastQC results.-f fastq: Informs FastQC that the input data is in FASTQ format.-q ${reads}: Specifies the input FASTQ file to be analyzed.-t ${task.cpus}: Utilizes the specified number of CPU cores for the analysis, which is determined by the configuration.
In summary, this Nextflow module, fastqc.nf wraps the execution of FastQC, ensuring that the FastQC results are organized and published to the specified output directory. It takes advantage of Nextflow's flexibility and parameterization to process multiple samples with different sample_id values, making it a reusable and efficient component of a larger NGS data analysis workflow.
- MultiQC Module
process MULTIQC {
publishDir "${params.output}/multiqc", mode: 'copy'
input:
path input_dir
output:
path '*'
script:
"""
multiqc .
"""
}
The provided code defines a Nextflow module called multiqc.nf that encapsulates the process of running MultiQC, a tool for summarizing and visualizing quality control results from multiple tools in a single report. Let's break down the code to understand its components:
Process Definition (MULTIQC):
- This module defines a process named "MULTIQC" using the
processkeyword. In Nextflow, a process represents a computational task or step in the workflow.
- This module defines a process named "MULTIQC" using the
publishDir "${params.output}/multiqc", mode: 'copy':- The
publishDirdirective specifies where the output files generated by this process should be published or copied. It uses${params.output}/multiqcas the destination directory, which typically points to the output directory defined in the workflow's configuration, with an additional subdirectory called "multiqc." This ensures that MultiQC output files will be collected and organized in a dedicated "multiqc" subdirectory within the specified output directory.
- The
Input Section:
The
inputsection defines the inputs required for this process:path input_dir: This input specification expects a single input path,input_dir, which represents the directory containing the output files from various quality control tools that MultiQC will analyze.
Output Section:
The
outputsection defines the expected output of this process:path '*': This output specification uses a wildcard to indicate that the process may produce multiple output files with various names. MultiQC generates a single report that summarizes the quality control results from the input directory.
Script Section:
The
scriptsection contains the actual shell script that will be executed by this process. It performs the following actions:- Executes the MultiQC tool with the command
multiqc .this command tells MultiQC to analyze the current directory (represented by.), which is where the input data from various quality control tools is expected to be located.
- Executes the MultiQC tool with the command
In summary, this Nextflow module, multiqc.nf wraps the execution of MultiQC, ensuring that MultiQC's summary report is organized and published to a dedicated "multiqc" subdirectory within the specified output directory. It allows for the consolidation of quality control results from various tools into a single, informative report, facilitating the assessment of data quality in NGS workflows.
D. Nextflow Workflow
nextflow.enable.dsl=2
include {FASTQC} from '/completepath/nextflow/pipelines/SequenceCleaning/modules/fastqc.nf'
include {MULTIQC} from '/completepath/nextflow/pipelines/SequenceCleaning/modules/multiqc.nf'
log.info """
S E Q U E N C E - C L E A N I N G - P I P E L I N E
===========================================================
Input : ${params.reads}
Output : ${params.output}
""".stripIndent()
Channel
.fromFilePairs(params.reads, checkIfExists: true)
.set { read_pairs_ch }
workflow {
FASTQC(read_pairs_ch)
MULTIQC(FASTQC.out.collect())
}
The provided Nextflow workflow script outlines a sequence cleaning pipeline using Nextflow DSL2. Let's break down the key elements and how the workflow operates:
nextflow.enable.dsl=2:- This line specifies that the Nextflow DSL2 (Domain-Specific Language 2) should be enabled for this workflow. DSL2 introduces enhancements and improvements for defining workflows in a more declarative and expressive manner.
include {FASTQC} from ...andinclude {MULTIQC} from ...:- These lines include two modules,
FASTQCandMULTIQC, from external files located at specific paths. These modules encapsulate the FastQC and MultiQC processes, making it easier to integrate them into the workflow.
- These lines include two modules,
log.info""" ... """.stripIndent():- This block generates a log message at the beginning of the workflow. It provides information about the workflow, including the input (
${params.reads}) and output (${params.output}) paths. The.stripIndent()function removes any leading indentation from the log message for cleaner formatting.
- This block generates a log message at the beginning of the workflow. It provides information about the workflow, including the input (
Channel.fromFilePairs(params.reads, checkIfExists: true):- This line creates a channel called
read_pairs_ch. A channel is a data structure in Nextflow that represents a stream of data. In this case, it reads data from file pairs specified by theparams.readsparameter. ThecheckIfExists: trueoption checks whether the input files exist before processing.
- This line creates a channel called
workflow { ... }:- The
workflowblock encapsulates the main workflow structure. Inside this block, you define how the processes (FASTQC and MULTIQC) are executed and connected.
- The
FASTQC(read_pairs_ch):- This line invokes the
FASTQCmodule and passes theread_pairs_chchannel as input. This means that the FASTQC process will operate on data streamed through theread_pairs_chchannel, which likely contains pairs of input FASTQ files.
- This line invokes the
MULTIQC(FASTQC.out.collect()):- This line invokes the
MULTIQCmodule and specifies that it should collect the output of the FASTQC process usingFASTQC.out.collect(). In other words, it gathers the results from all instances of the FASTQC process and feeds them into the MULTIQC process. MULTIQC will then generate a summary report based on the aggregated data from FASTQC.
- This line invokes the
In summary, this Nextflow workflow script sets up a sequence cleaning pipeline using the FASTQC and MULTIQC modules. It takes input data specified by params.reads, performs quality control with FASTQC on the input data in pairs, and then generates a summary report using MULTIQC. This modular and declarative approach allows for scalability and ease of integration of additional processes if needed.
E. Nextflow Run Pipeline
To run the sequence cleaning pipeline you've defined using Nextflow, you can use the following command:
nextflow run SequenceCleaning/SequenceCleaning.nf
Here's a breakdown of what this command does:
nextflow run: This is the standard command to execute a Nextflow workflow. It tells Nextflow to start running the workflow defined in the specified script.SequenceCleaning/SequenceCleaning.nf: This part of the command specifies the path to the Nextflow workflow script you want to run. In your case, the script is located in a folder called "SequenceCleaning," and the script itself is namedSequenceCleaning.nf
When you execute this command, Nextflow will start processing the workflow defined in SequenceCleaning.nf It will follow the instructions you've provided in the script, including any defined processes, inputs, outputs, and dependencies. This allows you to automate and manage the entire sequence cleaning process with ease.
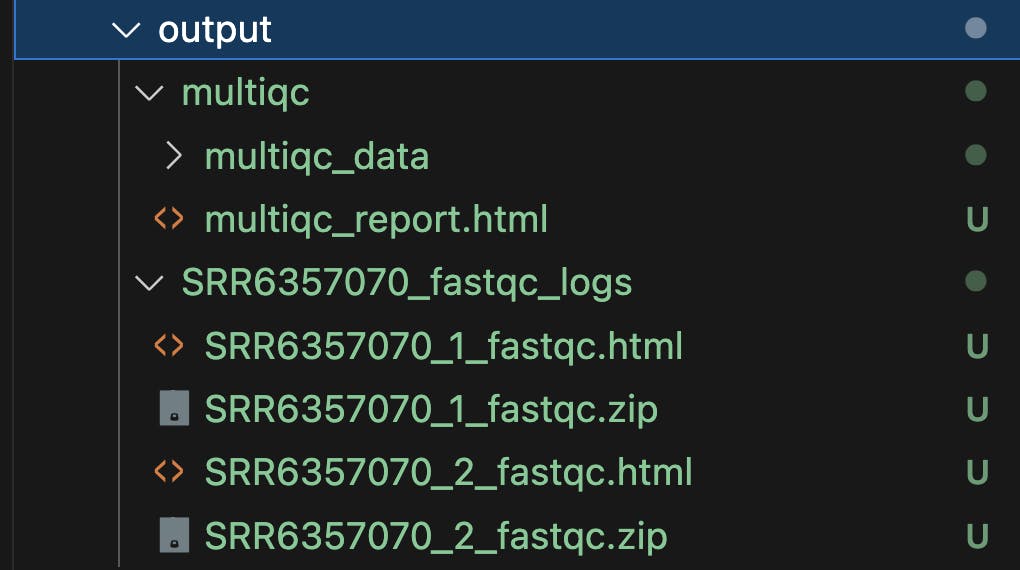
Pipeline Outputs
The described pipeline will produce FastQC reports, providing detailed insights into sequencing data quality, along with a MultiQC summary report that consolidates quality metrics from all samples for a comprehensive overview of data quality.
I hope you've enjoyed this article and picked up some cool stuff about Nextflow workflow manager! For more learning adventures, check out nf-core's tutorials, covering the basics and beyond:
Thank you for reading!
To gain a general understanding of next-generation sequencing (NGS) or to refresh your knowledge on the topic, please refer to the following article:
For more information on sequence cleaning and quality control of NGS data, please refer to the following article: